Diagnose
Die XLH-Diagnose basiert typischerweise auf klinischen und biochemischen Befunden in Kombination mit der Familienanamnese; Variationen im Krankheitsbild können jedoch zu einer verzögerten Diagnose oder Fehldiagnose führen.1,2
Mit Hilfe der Molekulargenetik kann eine Diagnose gestellt und bestimmt werden, ob eine XLH vererbt wird und welches Risiko für Familienmitglieder besteht.1
Klinisch
Bei Kindern:
Kinder mit XLH weisen in den ersten 1 bis 2 Lebensjahren typischerweise eine Verkrümmung der unteren Extremitäten, Wachstumsstörungen und Gangabweichungen auf. Die Diagnose wird manchmal jedoch erst nach dem 2. Lebensjahr oder sogar im Erwachsenenalter gestellt.1,3

Bei Erwachsenen:
Erwachsene Patienten weisen Gelenk- und Knochenschmerzen sowie Steifheit in Verbindung mit Arthrose und Enthesiopathie auf. Fast die Hälfte berichtet, eine Fraktur gehabt zu haben. Die Mehrheit der Erwachsenen mit XLH weist Kleinwuchs und Missbildung der unteren Extremitäten auf.2,4,5
Biochemisch
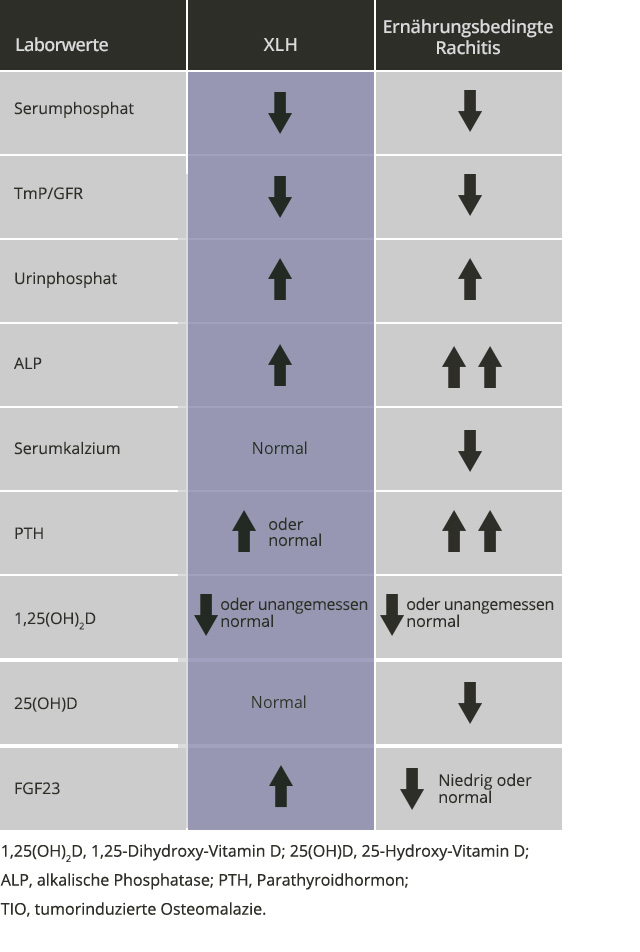
Wenn ein Patient klinische Merkmale aufweist, die denen der Rachitis ähneln, kann die Diagnose einer XLH mittels biochemischer Begutachtung gestellt werden
Die wichtigsten biochemischen Merkmale der XLH sind niedrige Serumphosphatspiegel, verringerte 1,25-Dihydroxyvitamin-D-Spiegel im Vergleich zur Serumphosphatkonzentration, ein verringertes Verhältnis zwischen der maximalen Phosphatreabsorption in den Nierentubuli und der glomerulären Filtrationsrate (TmP/GFR) sowie erhöhte FGF23-Serumspiegel.1,6,8
- Zu den zusätzlichen biochemischen Merkmalen der XLH gehören normale 25-Hydroxyvitamin-D-Spiegel, erhöhte Phosphorspiegel im Urin, erhöhte Spiegel der alkalischen Phosphatase und erhöhte oder normale Parathormonspiegel1,7,8
Phosphatreabsorption9–13
TmP/GFR ist das Verhältnis der maximalen Phosphatreabsorption in den Nierentubuli (TmP) zur glomerulären Filtrationsrate (GFR)

Familienanamnese
Kernpunkte:
- Die Begutachtung von gefährdeten Kindern ist gerechtfertigt, um eine frühe Diagnose und Behandlung zu gewährleisten, was nachweislich zu einer Verbesserung der klinischen Ergebnisse führt3
- Das Screening von Familienmitgliedern von XLH-Patienten kann helfen, bisher nicht diagnostizierte Personen zu identifizieren14
Stammbaumanalyse (engl. Pedigree Analysis)

Allerdings sind 20% bis 30% der Fälle spontan und haben daher keine Familienanamnese.15–17
1. Ruppe MD. X-linked hypophosphatemia. In: Pagon RA, Adam MP, Ardinger HH, et al, Hrsg. Gene Reviews. https://www.ncbi.nlm.nih.gov/books/NBK83985/. Abgerufen am 20. Oktober 2017. 2. Econs MJ, Samsa GP, Monger M, Drezner MK, Feussner JR. X-linked hypophosphatemic rickets: a disease often unknown to affected patients. Bone Miner. 1994;24(1):17-24. 3. Linglart A, Biosse-Duplan M, Briot K, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. 2014;3(1):R13-R30. 4. Skrinar A, Dvorak-Ewell M, Evins A, et al. The lifelong impact of X-linked hypophosphataemia: results from a burden of disease survey. J Endocr Soc. 2019;3(7):1321-1334. 5. Hardy DC, Murphy WA, Siegel BA, Reid IR, Whyte MP. X-linked hypophosphatemia in adults: prevalence of skeletal radiographic and scintigraphic features. Radiology. 1989;171(2):403-414. 6. Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician‘s guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26(7):1381-1388. 7. Santos F, Fuente R, Mejia N, Mantecon L, Gil-Peña H, Ordoñez FA. Hypophosphatemia and growth. Pediatr Nephrol. 2013;28(4):595-603. 8. Haffner D, Emma F, Eastwood DM, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15(7):435-455. 9. Payne RB. Renal tubular reabsorption of phosphate (TmP/GFR): indications and interpretation. Ann Clin Biochem. 1998;35(pt. 2):201-206. 10. Goldsweig BK, Carpenter TO. Hypophosphatemic rickets: lessons from disrupted FGF23 control of phosphorus homeostasis. Curr Osteoporos Rep. 2015;13(2):88-97. 11. Imel EA, Carpenter TO. A practical clinical approach to paediatric phosphate disorders. Endocr Dev. 2015;28:134-161. 12. Özkan B. Nutritional rickets. J Clin Res Pediatr Endocrinol. 2010;2(4):137-143. 13. Nield LS, Mahajan P, Joshi A, Kamat D. Rickets: not a disease of the past. Am Fam Physician. 2006;74(4):619-626. 14. Beck-Nielsen SS, Brusgaard K, Rasmussen LM, et al. Phenotype presentation of hypophosphatemic rickets in adults. Calcif Tissue Int. 2010;87(2):108-119. 15. Beck-Nielsen SS, Brixen K, Gram J, Brusgaard K. Mutational analysis of PHEX, FGF23, DMP1, SLC34A3 and CLCN5 in patients with hypophosphatemic rickets. J Hum Genet. 2012;57(7):453-458. 16. Gaucher C, Walrant-Debray O, Nguyen T-M, Esterle L, Garabédian M, Jehan F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet. 2009;125(4):401-411. 17. Whyte MP, Schranck FW, Armamento-Villareal R. X-linked hypophosphatemia: a search for gender, race, anticipation, or parent of origin effects on disease expression in children. J Clin Endocrinol Metab. 1996;81(11):4075-4080.