Clinical manifestations in adult patients with XLH
Clinical manifestations in adults with XLH arise from1,2:
- New and continuing symptoms as a result of ongoing, active disease
- Unresolved complications of XLH from childhood



Skeletal manifestations
Several types of fractures, including
can develop as a consequence of long-term weight bearing on weakened bones.1
These manifestations commonly result in spontaneous insufficiency fractures in the lower extremities and weight-bearing bones1
Nearly half of XLH patients reporting ever having fractures, with an average age at first fracture of 26.42
Skeletal manifestations continue into adulthood1,2,11
Ongoing, active disease in adults with XLH can cause several skeletal manifestations as a result of osteomalacia, or weakened bone.
Reported symptoms of osteomalacia include bone pain, muscle weakness and difficulty walking.
Frequency of skeletal and dental impairment in 232 adult XLH patients2

Bone repair and osteomalacia
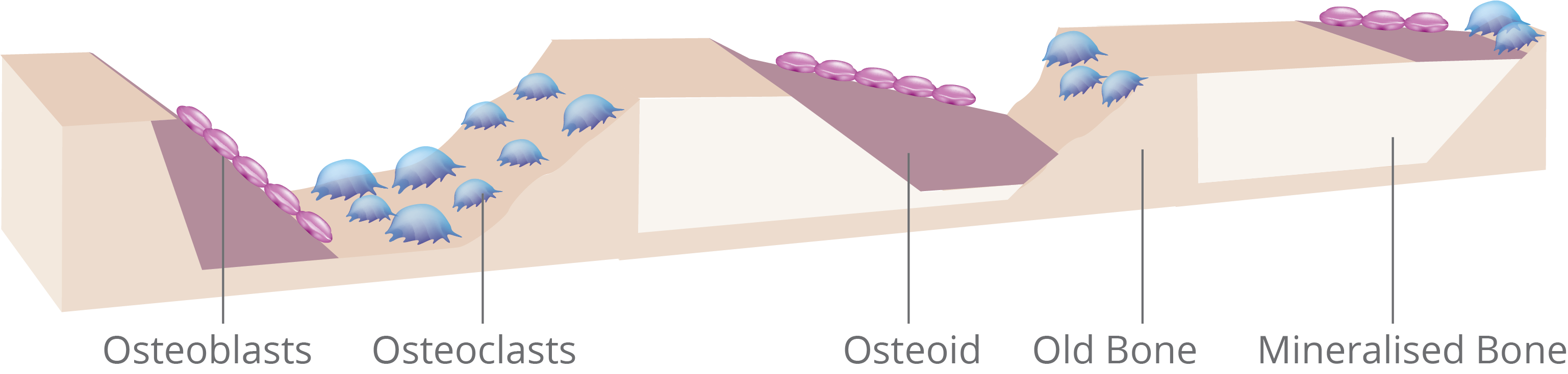
Normal Bone Formation7,12,13
- Osteoclasts bind and initiate bone remodelling to repair bone damage
- The mineralisation front should normally occupy >80% of the osteoid surface, with a slight decrease in older patientsa

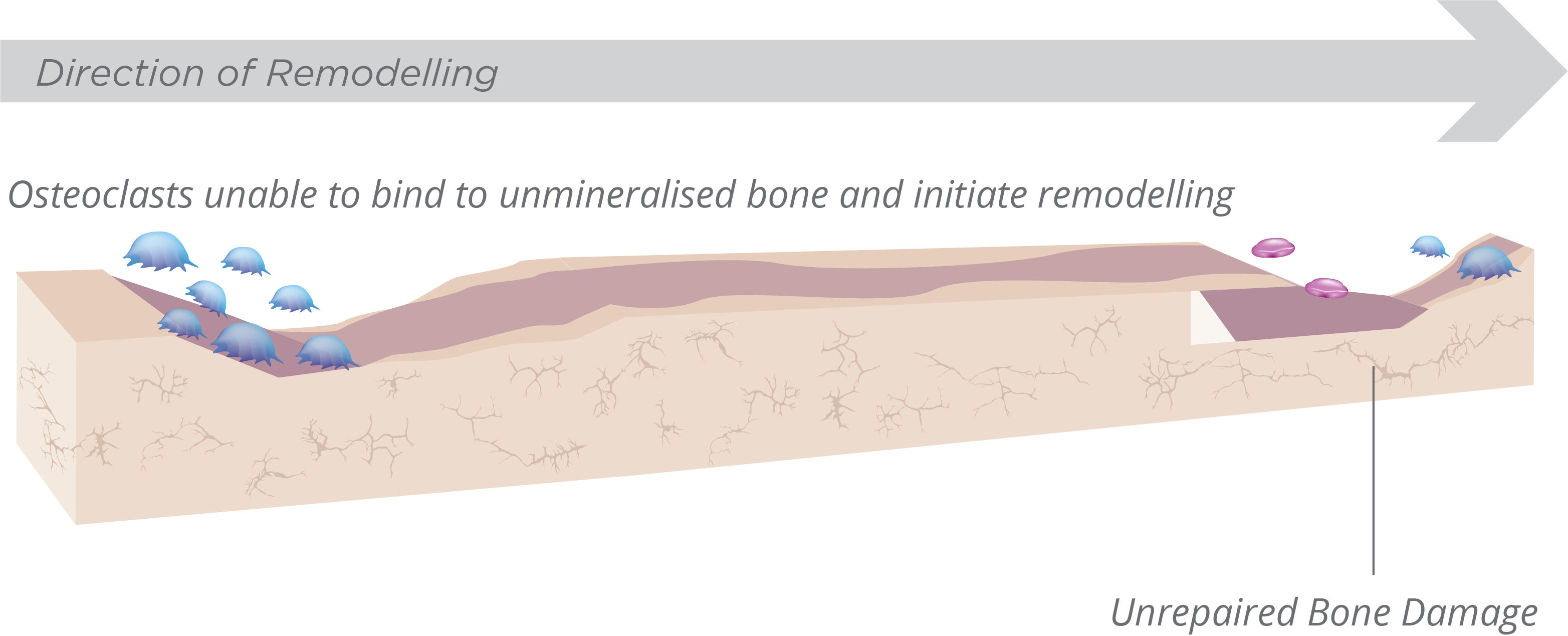
Osteomalacia7,12-14
- Osteoclasts have reduced ability to bind and initiate bone remodelling on undermineralised bone
- A mineralisation front of <20% represents a definite calcification defecta
Ongoing, active disease in adults with XLH can lead to impaired physical function.2
Adult patients with XLH most commonly report decreased range of motion, and gait disturbances, as well as pain and the need for walking assistance.2,8
Most frequently reported physical function limitations in 232 adult XLH patients2


Many adults with XLH have significantly impaired quality of life2
Pain, stiffness, and decreased physical function are the most common contributors to impaired quality of life in adults with symptomatic XLH.2
Complications associated with lower quality of life in adult XLH patients2

1. Linglart A, Biosse-Duplan M, Briot K, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. 2014;3(1):R13-R30. 2. Skrinar A, Dvorak-Ewell M, Evins A, et al. The lifelong impact of X-linked hypophosphataemia: results from a burden of disease survey. J Endocr Soc. 2019;3(7):1321-1334. 3. Veilleux LN, Cheung M, Ben Amor M, Rauch F. Abnormalities in muscle density and muscle function in hypophosphatemic rickets. J Clin Endocrinol Metab. 2012;97(8):E1492-E1498. 4. Looser zones. Radiopaedia Web site. https://radiopaedia.org/articles/looser-zones-1. Accessed October 9, 2017. 5. Jackson WPU, Dowdle E, Linder GC. Vitamin-D-resistant osteomalacia. Br Med J. 1958;1(5082):1269-1274. 6. Insufficiency fracture. Radiopaedia Web site. https://radiopaedia.org/articles/insufficiency-fracture. Accessed October 9, 2017. 7. Martin A, Quarles LD. Evidence for FGF23 involvement in a bone-kidney axis regulating bone mineralization and systemic phosphate and vitamin D homeostasis. Adv Exp Med Biol. 2012;728:65-83. 8. Che H, Roux C, Etcheto A, et al. Impaired quality of life in adults with X-linked hypophosphatemia and skeletal symptoms. Eur J Endocrinol. 2016;174(3):325-333. 9. Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician's guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26(7):1381-1388. 10. Econs MJ, Samsa GP, Monger M, Drezner MK, Feussner JR. X-linked hypophosphatemic rickets: a disease often unknown to affected patients. Bone Miner. 1994;24(1):17-24. 11. Reid IR, Hardy DC, Murphy WA, Teitelbaum SL, Bergfeld MA, Whyte MP. X-linked hypophosphatemia: a clinical, biochemical, and histopathologic assessment of morbidity in adults. Medicine (Baltimore). 1989;68(6):336-352. 12. Revell PA. Histomorphometry of bone. J Clin Pathol. 1983;36(12):1323-1331. 13. Miller PD. Renal bone disease. In: Orwoll ES, ed. Atlas of Osteoporosis. 3rd ed. Current Medicine Group; 2009. 14. Chambers TJ, Thomson BM, Fuller K. Effect of substrate composition on bone resorption by rabbit osteoclasts. J Cell Sci. 1984;70:61-71.